
Contact
- yph@gate.sinica.edu.tw
- (L) 886-2-2789-9311
- N514, Institute of Molecular Biology, Academia Sinica
Research
Neuronal morphogenesis, neurodevelopmental disorders, and neurodegeneration
It is well known that the defects in neural development may result in complex neurodevelopmental disorders, including mental retardation, autism, ADHD, and schizophrenia. We are interested in the molecular mechanisms underlying neuronal morphogenesis, particularly synapse formation, and how the defects in neuronal development result in cognitive and psychiatric disorders. Both cultured hippocampal neurons and mouse genetic models are used in my lab to explore the mechanism of neuronal morphogenesis and the biological significance of genes of interests.
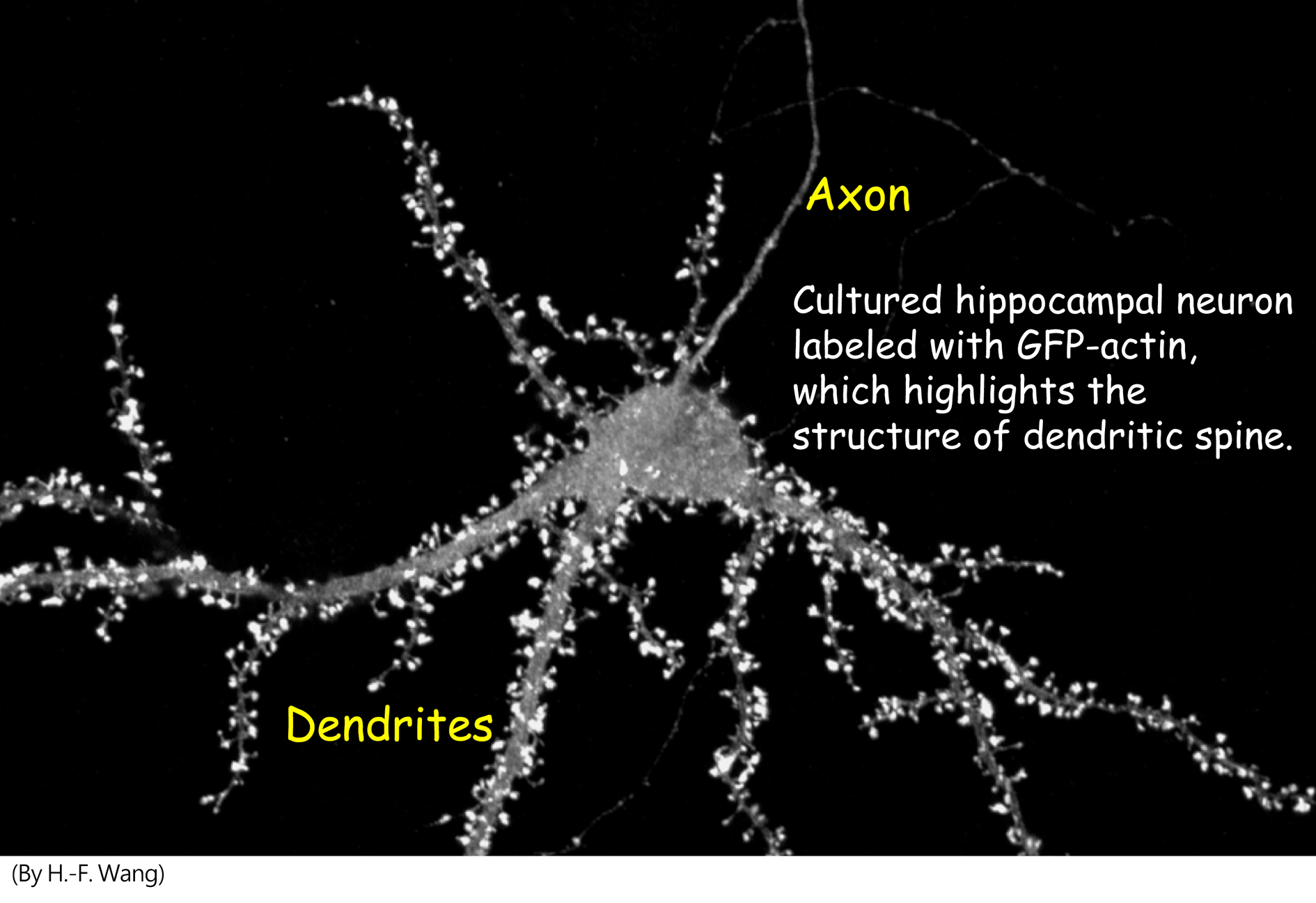
I. CASK and X-linked mental retardation
CASK, a member of membrane associated guanylate kinase (MAGUK) family, has been identified as a causative gene of X-linked mental retardation. It functions as a multidomain scaffold protein to interact with more than twenty of binding partners. The CASK protein network that we have been focusing on is summarized in the cartoon. Briefly, we showed that CASK enters the nuclei of neurons and interacts with several nuclear proteins, including T-box transcription factor Tbr1 (T-brain-1), nucleosome assembly protein CINAP (CASK interacting nucleosome assembly protein), and zinc finger transcription factor Bcl11A (B cell lymphoma 11A). Tbr1 and CINAP form a complex with CASK and regulate expression of NMDAR2b gene. For Bcl11A, we found that it negatively influences axonal outgrowth and branching. Overexpression of CASK further promotes the activities of Bcl11A in regulation of neurite outgrowth and branching. It is not clear whether Bcl11A also forms complex with Tbr1 and CINPA through the interaction with CASK.
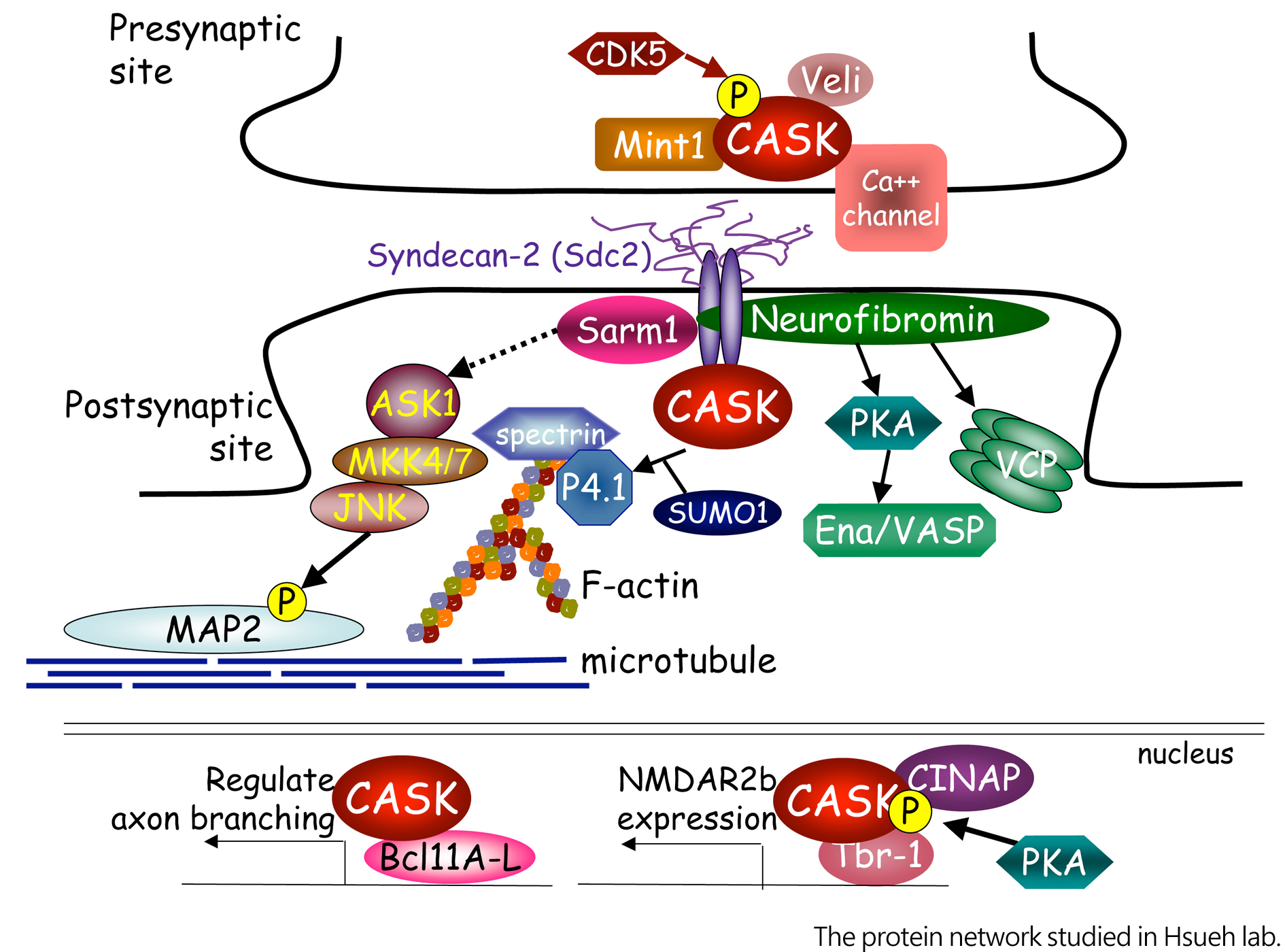
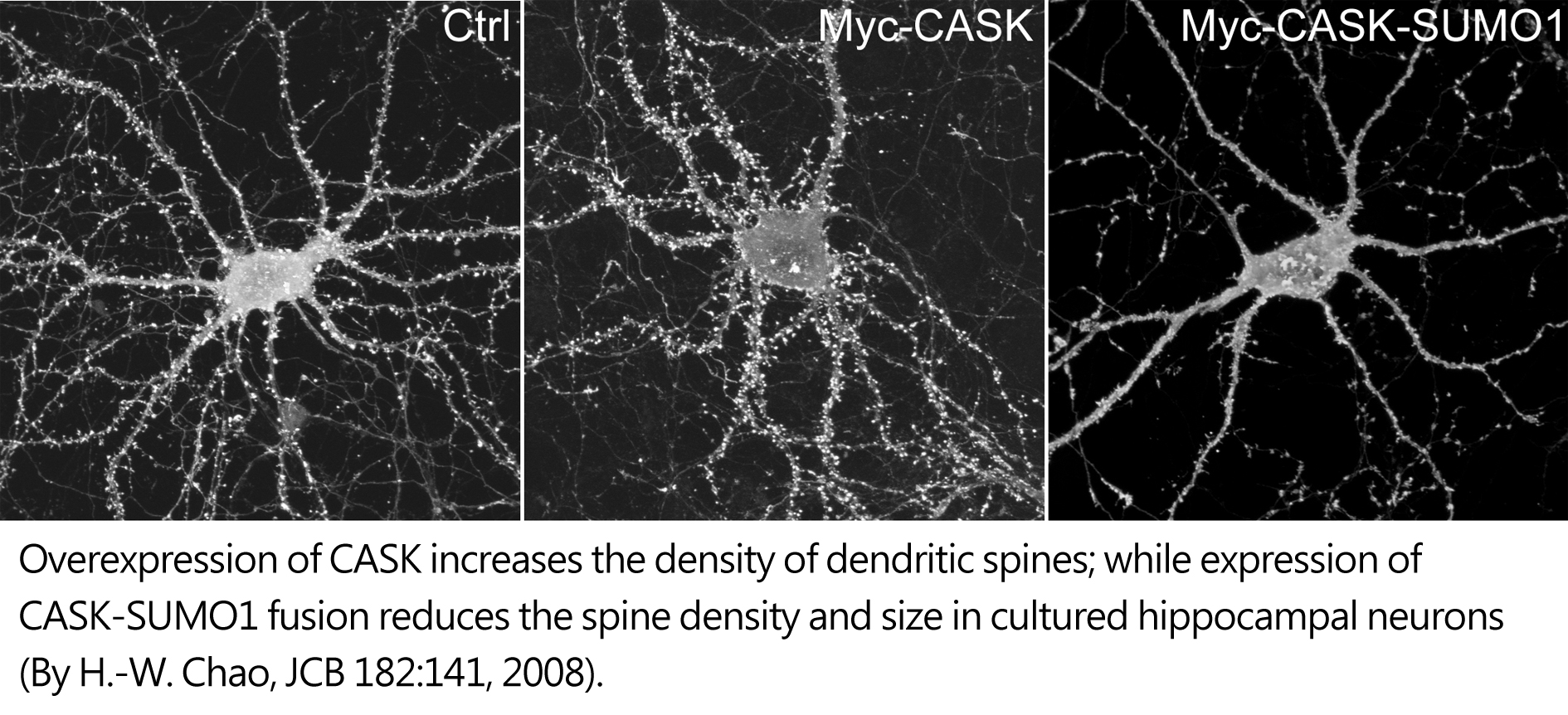
Like other MAGUK proteins, CASK is also present at synapses, where it interacts with neurexin at the presynaptic site and syndecan-2 at the postsynaptic site. Both neurexin and syndecan-2 are transmembrane proteins involved in cell adhesion. CASK had been therefore suggested in synaptic interaction. Using RNAi knockdown and domain deletion analysis, we showed that CASK is required for dendritic spine formation. CASK links transmembrane proteoglycan syndecan-2 to protein 4.1-F-actin cytoskeletons and thus stabilizes dendritic spines. This function is regulated by protein SUMOylation. CASK SUMOylation reduces the interaction between CASK and protein 4.1 and destabilizes dendritic spines. At presynaptic sites, plasma membrane association of CASK is controlled by CDK5 phosphorylation, which is critical for formation of presynaptic boutons. Thus, the roles of CASK in synapse formation are regulated by SUMOylation and CDK5 phosphorylation.
Our studies reveal the functions of CASK in regulation of dendritic spine formation and NMDAR2b expression, which provide the molecular mechanisms underlying the roles of CASK in mental retardation.
II. Neuronal morphogenesis, Innate immunity, and autism
Syndecan-2, a CASK interacting transmembrane heparan sulfate proteoglycan, is highly concentrated at synapses in mature neurons. Overexpression of syndecan-2 accelerates dendritic spine formation and dendritic outgrowth. For spinogenesis, syndecan-2 promotes filopodia formation through the interaction with neurofibromin, a protein product of Neurofibromatosis Type I (NF1) gene. Neurofibromin increases the PKA activity and thus promotes Ena/VASP phosphorylation and bundle formation of F-actin. The filopodia are then transformed to mature dendritic spines, a process requiring the interaction between the C-terminal tail of syndecan-2 and PDZ proteins, such as CASK. Our study has revealed the molecular regulation of syndecan-2 in spinogenesis. Interestingly, syndecan-2 has been reported to associate with autism. The role of syndecan-2 in spinogenesis echoes a current hypothesis that synapses are the primary targets affected in patients with autism.
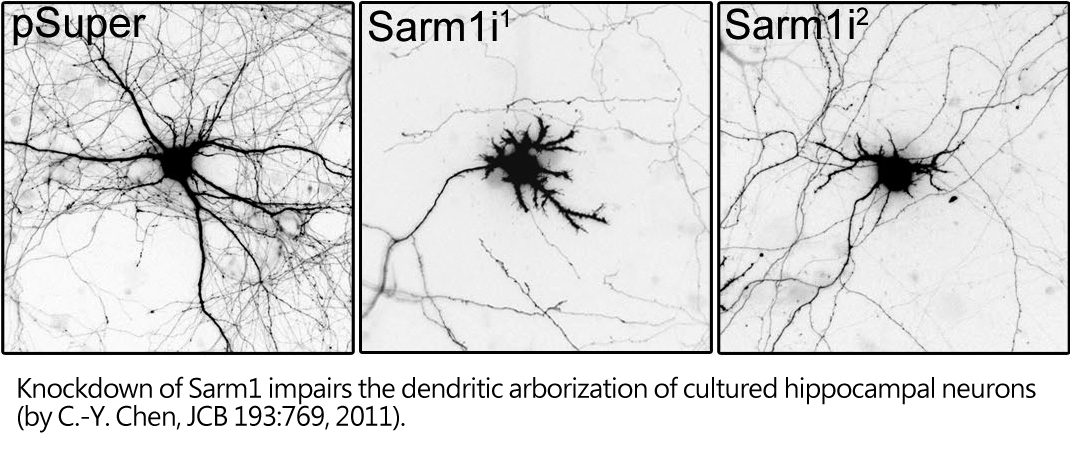
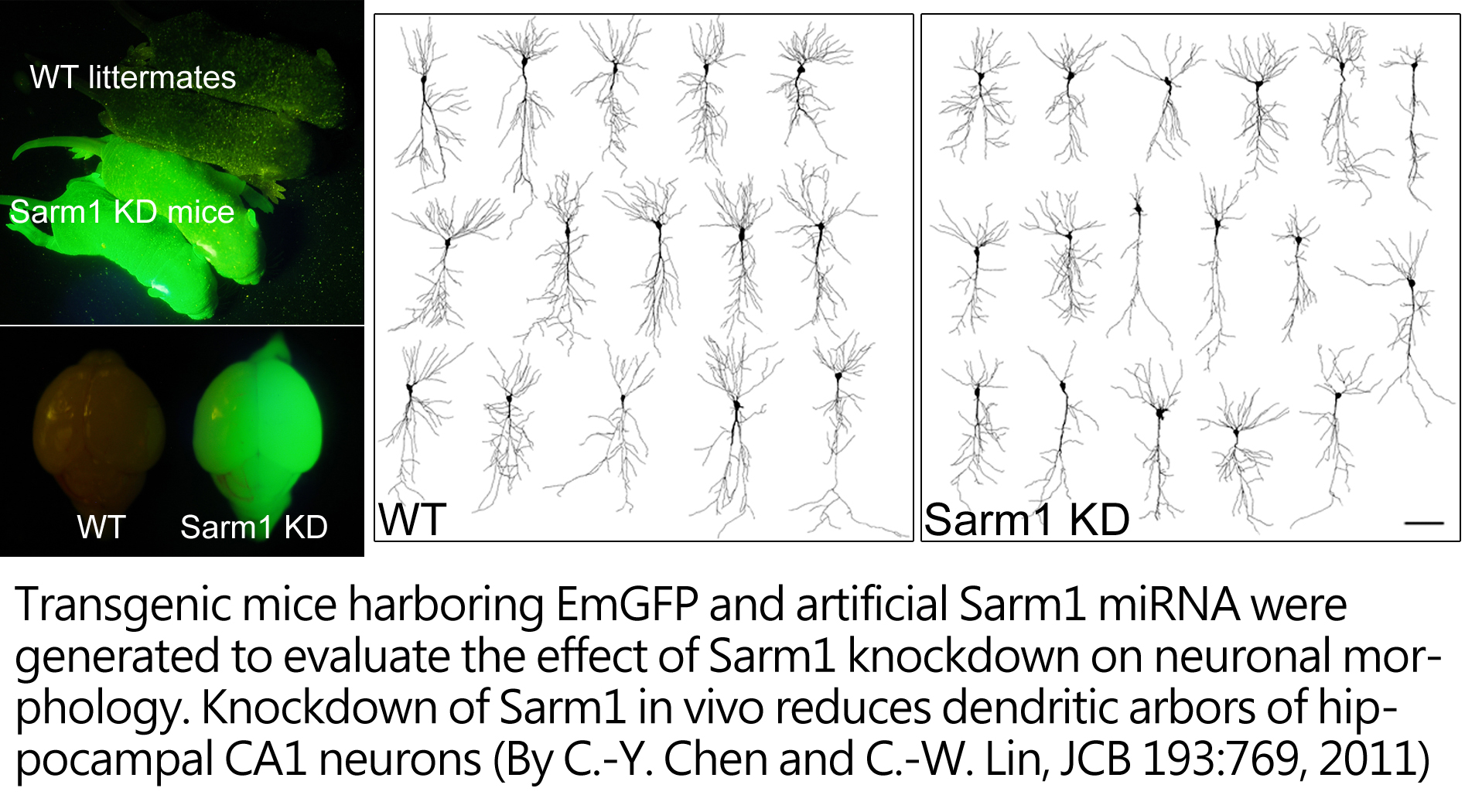
In addition to synapse formation, syndecan-2 also modulates dendritic arborization. We found that Sarm1 (sterile alpha and TIR motif containing 1) plays a critical regulatory role in dendritic outgrowth. Using proteomic approaches, Sarm1 was identified as interacting protein of syndecan-2. Sarm1 knockdown impairs both intrinsic dendritic outgrowth and the outgorwth induced by sydenca-2 overexpression. The analysis of Sarm1 knockdown transgenic mice also supports a role of Samr1 in dendritic arborization. With the Sarm1 mutant mice in our hands, we further analyzed whether reduction of Sarm1 expression has an impact on cognition.
Although Sarm1 is expressed at the highest levels in brain, Sarm1 was originally identified as a negative regulator in the signal pathways of TLR3 and TLR4. Since neonatal infection increases the risk to develop psychiatric disorders, such as autism and schizophrenia, we are also in progress to explore the role of Sarm1 in TLR3-dependent psychiatric disorder and the potential role of other TLRs in neuronal development.
III. A link between neurodevelopmental disorder and neurodegeneration — the interaction of neurofibromin and VCP/p97.
Neurofibromatosis type I (NF1) is an autosomal dominant genetic disorder affecting about one in 3,500 individuals. There are four major features of NF1, i.e. Café-au-lait spots, peripheral neurofibromas, skinfold freckling, and Lisch nodules. In addition, many other features are frequently found in patients with NF1, including cognitive deficits as well as skeletal lesions and malformations. Children with NF1 are frequently associated with learning difficulty and are more susceptible to autism. Our finding about the role of neurofibromin in dendritic spinogenesis provides an explanation why patients with NF1 suffer from learning disability and autism. We are now further exploring the molecular regulation and pathway underlying the role of neurofibromin in neuronal morphogenesis. Although we found that PKA is a critical downstream effector of neurofibromin in regulation of dendritic spine formation, activation of PKA alone is not sufficient for initiation of spinogenesis, suggesting that multiple pathways downstream of neurofibromin are involved in spinogenesis. Using proteomic approaches, we identified VCP/p97 (valosin containing protein) as a novel neurofibromin interacting protein. The experiments combining cultured neurons and mouse genetic model suggest that VCP/p97 acts downstream of neurofibromin in controlling neuronal morphogenesis. VCP/p97 is the causative gene of IBMPFD (Inclusion body myopathy with early-onset Paget disease and frontotemporal dementia). The role of VCP/p97 in neuronal morphogenesis provides the potential explanation why VCP/p97 mutations result in dementia. This is the first evidence linking Neurofibromatosis type I and IBMPFD. We are now continuously establishing and using mouse genetic models to further elucidate the detail molecular regulation and pathogenesis.
IV. CTTNBP2 and dendritic spine formation.
CTTNBP2 (Cortactin binding protein 2) is a brain predominant protein, which interacts with cortactin. Cortactin, a ubiquitous protein, regulates F-actin branching and stability. Although cortactin is not neuron-specific, it is highly enriched at dendritic spines and has been suggested to regulate neuronal activity-depdendent remodeling. Using immunostaining and FRAP analysis, we investigate whether CTTNBP2 is also involved in dendritic spinogenesis and whether CTTNBP2 controls the function of cortactin to achieve a neuron-specific function, namely dendritic spine formation. Since CTTNBP2 gene is located at chromosome 7q31, an autism candidate region, it is intriguing to explore whether CTTNBP2 also associates with autism.